
Alexa Woodward
Gene-Interaction-Sensitive Enrichment Analysis in Congenital Heart Disease

Abstract
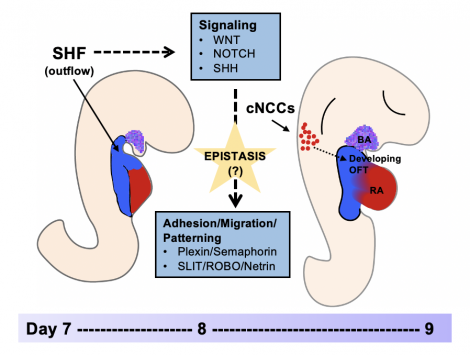
Background: Gene set enrichment analysis (GSEA) uses gene-level univariate associations to identify gene set-phenotype associations for hypothesis generation and interpretation. We propose that GSEA can be extended to incorporate SNP and gene-level interactions. Relief-based algorithms (RBAs) are feature importance scoring and selection methods that uniquely capture both main effects and epistatic interactions with computational efficiency. We hypothesize that using RBA scores within GSEA will empower discoveries and interpretations that account for interactions.
Methods: We utilized GWAS data from two cohorts with conotruncal defects (CTDs) as our discovery and replication datasets. We used PLINK to obtain chi-square statistics from a standard case-control association analysis. As our novel alternative, we applied the two Relief-based feature selection algorithms that detect both univariate and interaction effects (MultiSURF) or exclusively detects interactions (MultiSURF*) to calculate feature scores. GSEA was then conducted for each algorithm using respective ranked gene lists, along with leading-edge and correlation analyses..
Results and Conclusions: Both Relief-based approaches to GSEA captured more relevant and significant gene ontology (GO) terms compared to the univariate GSEA. Key GO terms and themes of interest from the Relief-based approach include cell adhesion, migration, and signaling. A leading-edge analysis highlighted semaphorins and their receptors, the Slit-Robo pathway, and other genes with roles in outflow tract development. By accounting for potential interactions, we replicated univariate findings and identified additional and more robust support for the role of the secondary heart field and cardiac neural crest cell migration in the development of CTDs.
Keywords
GSEA, Relief-based algorithms, epistasis, GWAS, CHD, conotruncal defectsCommenting is now closed.
About Us
To understand health and disease today, we need new thinking and novel science —the kind we create when multiple disciplines work together from the ground up. That is why this department has put forward a bold vision in population-health science: a single academic home for biostatistics, epidemiology and informatics.
© 2023 Trustees of the University of Pennsylvania. All rights reserved.. | Disclaimer